Introdução
As vasculites associadas ao anticorpo citoplasmático antineutrófilo (ANCA) (VAA) incluem granulomatose com poliangiite (GPA), poliangiite microscópica (PAM) e granulomatose eosinofílica com poliangiite (GEPA).
VAA representam um subgrupo dentro do espectro de vasculite sistêmica primária.
A GPA (antigamente chamada de granulomatose de Wegener) é caracterizada histologicamente por inflamação granulomatosa necrosante, além de vasculite. A incidência mundial anual de GPA é estimada em 10-20 casos por um milhão. Tem um pico de incidência entre 64 e 75 anos de idade e estudos recentes não mostraram predileção por sexo. É comumente relatada em brancos, embora possa ser observada em todos os grupos raciais e étnicos.
PAM é caracterizado histologicamente por vasculite sem inflamação granulomatosa. A poliangeíte microscópica tem uma incidência relatada de 2,7 a 94 por 1 milhão. É mais comum na população branca do que na negra, com idade média de início é de 50 anos, sendo mais prevalente em homens.
GEPA é caracterizada histologicamente por infiltração tecidual eosinofílica além de vasculite. A incidência da EGPA varia entre 0,5 e 4,2 casos por milhão de pessoas por ano e sua prevalência entre 10 e 14 casos por milhão de habitantes globalmente. A frequência da doença é comparável em homens e mulheres, e a idade média no momento do diagnóstico é de aproximadamente 50 anos.
Os regimes de tratamento atuais reverteram o mau prognóstico da doença (antes dos tratamentos a sobrevida média de pacientes com GPA era de aproximadamente 5 meses), mas os tratamentos ainda estão associados à toxicidade.
Definições:
Doença ativa: presença de sinais, sintomas ou outras características típicas (como glomerulonefrite ou nódulos pulmonares) de VAA ativa.
Remissão: ausência de sinais, sintomas ou outras características típicas de VAA ativa com ou sem terapia imunossupressora.
Remissão sustentada: ausência de sinais, sintomas ou outras características típicas de VAA ativa durante um período de tempo definido, com ou sem terapia imunossupressora.
Resposta: redução ≥50% do escore de atividade da doença e ausência de novas manifestações.
Recaída: recorrência de VAA ativa após um período de remissão.
Refratários: sinais, sintomas ou outras características inalterados ou aumentados de AAV ativa após um período de terapia de indução padrão. Danos, infecções, efeitos colaterais do tratamento ou comorbidades como possíveis causas das manifestações persistentes ou agravadas da doença precisam ser descartados.
Dividir os pacientes entre “grave” e “não grave” pode favorecer um grupo a receber tratamento menos intenso, e ainda assim correr o risco de desenvolver manifestações que ameaçam os órgãos ou ameaçam a vida.
Exemplos de manifestações potencialmente fatais: glomerulonefrite, hemorragia pulmonar, envolvimento meníngeo, envolvimento do sistema nervoso central, doença retro-orbital, envolvimento cardíaco, envolvimento mesentérico, moneurite múltipla.
Exemplo de manifestações que não representam risco para vida: doença nasal e paranasal (sem envolvimento ósseo/colapso de cartilagem/olfatório/surdez), envolvimento cutâneo sem ulceração, miosite, nódulos pulmonares não cavitados, episclerite.
Fisiopatologia
GEPA
A patogênese é impulsionada por fatores genéticos e ambientais
Estudos genéticos destacaram associações entre HLA-DQ e GEPA positivo para MPO-ANCA, enquanto o GEPA negativo para ANCA está associado principalmente a variantes genéticas envolvidas nas respostas da mucosa e na biologia dos eosinófilos, como GPA33 e IL5.
Entre os fatores ambientais, a exposição à sílica, solventes orgânicos e agricultura foi associada a um risco aumentado de GEPA, enquanto o tabagismo foi associado a um risco menor.
Vários tipos celulares participam da imunopatogênese da doença. Os eosinófilos são claramente centrais e provavelmente medeiam danos teciduais, um conceito apoiado pela evidência de que direcionar a IL-5 (por exemplo, usando mepolizumab).
As células T CD4 orquestram a resposta imune adaptativa e são polarizadas em direção a um fenótipo T helper (TH2), que aumenta as reações eosinofílicas; entretanto, as células TH1 e TH17 também podem ter um papel, especialmente na vasculite e na formação de granulomas.
As respostas humorais e de células B também estão desreguladas na GEPA, além da produção de ANCA, a produção aumentada de IgG4 é uma característica.
GPA
A causa exata do GPA não é bem compreendida. A etiopatogenia tem sido creditada a anticorpos citoplasmáticos anti-neutrofílicos (ANCA). Várias interações complexas envolvendo genética e microorganismos foram implicadas na patogênese.
Respostas imunorregulatórias defeituosas a insultos ambientais, como infecção ou autoantígenos, levam à produção excessiva de citocinas Th1 e Th17 (interleucina 17, fator de necrose tumoral e interferon-gama), que por sua vez pode levar ao desenvolvimento de uma lesão vascular granulomatosa inflamatória.
O ANCA ativa os neutrófilos, o que leva ao aumento da adesão ao endotélio e induz a sua desgranulação que pode danificar as células endoteliais.
As associações genéticas no GPA incluem: alelo defeituoso para alfa 1 antitripsina; proteína 4 associada a linfócitos T citotóxicos (CTLA-4), que está envolvida na ativação de células T; gene da proteinase 3 (PRTN 3); complexo principal de histocompatibilidade, classe II, gene DP alfa 1 (HLA-DP); certos tipos de receptor gama FC III b na superfície de neutrófilos e macrócitos/monócitos.
De associações infecciosas: colonização por Staphylococcus aureus, vírus da hepatite C (HCV), citomegalovírus (CMV), vírus Epstein-Barr (EBV) e parvovírus.
Medicamentos: vários medicamentos como hidralazina, fenitoína, medicamentos antitireoidianos, sulfassalazina e alopurinol foram implicados.
A formação dos granulomas no GPA começa com a formação de microabscessos neutrofílicos. Os granulomas no GPA resultam em oclusão parcial ou total dos vasos sanguíneos. Os granulomas na GPA não são bem formados, ao contrário dos da sarcoidose ou da tuberculose, e consistem em células gigantes rodeadas por plasmócitos, linfócitos e células dendríticas. Essas células podem danificar a submucosa e penetrar nos tecidos circundantes, na cartilagem ou nos ossos, resultando em necrose e deformidades permanentes.
PAM
A etiopatogenia da poliangeíte microscópica (MPA) e outras vasculites relacionadas tem sido amplamente atribuída a anticorpos citoplasmáticos antineutrófilos ou ANCA.
Estes anticorpos reagem contra grânulos primários presentes em neutrófilos e monócitos. A formação desses anticorpos foi considerada um processo de duas etapas. A primeira etapa envolve a exposição de neutrófilos a citocinas inflamatórias, levando à exposição superficial de antígenos criptogênicos como mieloperoxidase ou MPO. Em seguida, fatores predisponentes genéticos, ambientais e outros resultam na produção de MPO-ANCA. Na segunda etapa, estes MPO-ANCA causam danos à vasculatura do hospedeiro ao reagir com neutrófilos aos receptores endoteliais.
Apenas 70% dos casos de PAM apresentam ANCA no momento do diagnóstico, e a maioria dos casos de PAM limitado não apresenta ANCA. Isto levou ao entendimento de que outros fatores também podem desempenhar um papel na sua etiopatogenia.
Infecciosas: Existe uma sobreposição considerável na apresentação clínica de vários processos infecciosos e PAM que levam a esta implicação.
Medicamentos: observou-se que vários medicamentos como hidralazina, tionamidas, sulfassalazina e minociclina, entre outros, estão associados à incidência de vasculite associada a ANCA.
Genéticos: HLA-DP , HLA-DR3 e alfa-1 antitripsina.
Ambiental: sílica.
A poliangeíte microscópica está associada ao MPO-ANCA em 58% dos casos e ao PR3-ANCA em 26% dos casos.
A PAM é caracterizada por vasculite pauci-imune, necrosante e de pequenos vasos, sem evidência clínica ou patológica de inflamação granulomatosa.
Manifestações Clínicas
Granulomatose eosinofílica com poliangiite
Anteriormente conhecida como síndrome de Churg-Strauss, é uma forma de vasculite que é histologicamente definida por inflamação granulomatosa necrosante, rica em eosinófilos, envolvendo principalmente o trato respiratório, juntamente com vasculite necrosante de vasos de pequeno à médio porte.
Evolui através de três fases diferentes: uma fase prodrômica ‘alérgica’, que pode durar vários anos e é marcada por asma e rinossinusite crônica; uma fase eosinofílica, durante a qual aparecem eosinofilia e envolvimento de órgãos-alvo; e uma fase vasculítica, caracterizada por manifestações clínicas devido a vasculite de pequenos vasos (por exemplo, mononeurite múltipla e glomerulonefrite). Entretanto, essas fases muitas vezes se sobrepõem, não necessariamente se desenvolvem na sequência citada e alguns pacientes não manifestam complicações vasculíticas.
Os pacientes apresentam clínica de asma (asma do adulto), com sinias de doença pulmonar obstrutiva (ocorrendo em até 90% dos pacientes), raramente apresenta exacerbações sazonais e tende a piorar com o tempo.
Acometimento de nariz, garganta e ouvido em até 60-80% dos casos.
Sinusopatia, otite média, pólipos nasais (geralmente recidivantes), quando realizado a biópsia com presença de infiltrado eosinofílico (com ou sem granulomas).
Sistema nervoso periférico com neuropatia (50-70%, moneurite múltipla). Os nervos fibulares comuns e poplíteos internos são mais comumente envolvidos. Pode ocrrer vasculite do SNC, causando acidente vascular cerebral ou hemorragias. As paralisias dos nervos cranianos são incomuns, mas o envolvimento dos nervos cranianos 2, 3, 6 e 8 foi descrito. Pode ocorrer neuropatia optica isquêmica.
No hemograma com eosinofília periférica (mais que 10% ou 1000-1500 eosinófilos), pode ser mascarado pelo uso prévio de corticoide.
ANCAs são detectados em aproximadamente 40% a 60% (pANCA, MPO-ANCA). Características de vasculite, particularmente glomerulonefrite, neuropatia periférica e púrpura, ocorrem mais frequentemente em pacientes ANCA-positivos.Características eosinofílicas, como envolvimento cardíaco e gastroenterite, são mais frequentes em pacientes ANCA-negativos.
O envolvimento cardíaco pode estar presente em 62% dos casos, embora se manifeste sintomaticamente em apenas 26%. É causada tanto por mediadores liberados por eosinófilos ativados quanto por lesões de vasculite no miocárdio e nas artérias coronárias. A miocardite, por sua vez, leva à fibrose pós-inflamatória e à cardiomiopatia restritiva, seguida de insuficiência cardíaca congestiva. O espectro de manifestações clínicas varia de doença arterial coronariana, arritmias primárias, cardiomiopatia, pericardite e miocardite constritiva aguda e derrame pericárdico eosinofílico. Realizar ecocardiograma em todo paciente com GEPA. As alterações cardíacas estão associadas a mau prognóstico e alta mortalidade se não tratadas.
Pulmonar pode levar a presença de infiltrado pulmonar (40–50%), muitas vezes são múltiplos e migratórios e respondem ao tratamento com glicocorticoides sistêmicos. Pode ocorrer hemorragia alveolar.
Lesões cutâneas também são frequentes, mas bastante heterogêneas, sendo a púrpura palpável a lesão mais específica da vasculite.
O envolvimento renal é observado em 25% dos pacientes e é menos comum do que outras vasculites anca associadas. Glomerulonefrite revelada por proteinúria, hematúria e/ou graus variados de insuficiência renal. A manifestação mais comum é a glomerulonefrite crescente necrosante.
Gastrointestinal: a gastroenterite eosinofílica e a vasculite mesentérica muitas vezes parecem coincidir no que diz respeito ao envolvimento do trato gastrointestinal. Eles resultam em sintomas inespecíficos de dor abdominal, náuseas, vômitos e diarreia, até complicações mais graves, como sangramento ou obstrução intestinal causada por massas nodulares submucosas. A vasculite mesentérica predispõe à isquemia intestinal, ulceração da mucosa e até mesmo perfuração. O envolvimento seroso pode causar ascite eosinofílica e peritonite. As manifestações raras incluem colecistite acalculosa necrosante, pancreatite e doença hepática eosinofílica.
Constitucionais: fadiga, perda de peso, mialgia e artralgia.
Granulomatose com poliangiíte
A granulomatose com poliangiíte comumente envolve uma tríade de a) trato respiratório superior (sinusite, rinite com crostas, deformidade do nariz em sela, otite média, mastoidite, perda auditiva) e trato respiratório inferior (nódulos pulmonares, hemorragia alveolar), b) vasculite sistêmica e c) envolvimento renal (glomerulonefrite).
O ANCA dirigido contra a proteinase 3 pode ser encontrado em 80% dos pacientes com GPA
Tratato respiratório superior: 90% dos pacientes apresentam esse envolvimento. Dor nasal e seios da face, congestão nasal, secreção nasal purulenta/sanguinolenta, ulcerações nasais, epistaxe e otite média, hiperplasia gengival. A presença de sinais clínicos de sinusite, rinite com crostas, otite média, mastoidite e perda auditiva deve alertar para GPA. A inflamação nasal pode causar perfuração septal ou colapso da ponte nasal, causando deformidade do nariz em sela.
Trato respiratório inferior: tosse, hemoptise, dispneia, dor torácica pleurítica e obstrução traqueal. Inicialmente, quase 50% dos pacientes apresentam infiltrados pulmonares bilaterais ou unilaterais. Nódulos pulmonares e cavitação também são observados. Derrame pleural também foi relatado, assim como hemorragia alveolar.
A estenose subglótica e a estenose brônquica são uma complicação potencialmente grave da GPA. Os sintomas podem variar desde inicialmente assintomáticos até o desenvolvimento de rouquidão, tosse, sibílos ou estridor.
Renal: a manifestação mais comum é a glomerulonefrite crescente rapidamente progressiva, levando à doença renal crônica ou à doença renal em estágio terminal.
Gastrointestinal: vasculite mesentérica, dor abdominal, peritonite.
Ocular: esclerite e conjuntivite são mais comumente observadas. A esclerite pode causar esclerite anterior necrotisante. A ceratite ulcerativa periférica (PUK) é a complicação corneana mais significativa do GPA. Outras manifestações incluem episclerite, uveíte, vasculite retiniana, trombose venosa ou arterial da retina. Em 10% a 15% dos pacientes podem ocorrer massas orbitais na região retrobulbar, denominadas pseudotumores, podendo causar diplopia, proptose ou perda de visão. A obstrução do ducto nasolacrimal é frequentemente observada na GPA.
Ouvido: ambas as categorias de doenças do ouvido – perda auditiva condutiva e neurossensorial são típicas da doença. A perda auditiva condutiva devido à disfunção da tuba auditiva secundária à doença nasofaríngea é observada em muitos casos de GPA. Perda auditiva neurossensorial e disfunção vestibular são observadas em alguns indivíduos. O envolvimento do ouvido médio, incluindo otite média serosa e mastoidite, também é observado.
Cutâneo: púrpura e nódulos cutâneos
Sistema nervoso: as neuropatias periféricas as mais comuns, pode levar à mononeurite múltipla. Neuropatias cranianas, paquimeningite, convulsões e cerebrite também foram relatadas.
Cardíaco: o envolvimento cardíaco é menos comum e envolve lesões ou insuficiência valvar, pericardite e arterite coronária.
Na histologia das biópsias:
Pulmão: Granulomas circundados por histiócitos em paliçada e células gigantes com necrose central. Isso leva à liquefação/necrose coagulativa nos pulmões com eosinófilos profusos e células gigantes multinucleadas. É observada angiite de artérias e veias por neutrófilos, plasmócitos e eosinófilos, escassos linfócitos e plasmócitos.
Rim: A glomerulonefrite necrosante focal é observada frequentemente com crescentes celulares e trombose glomerular, pauci-imune. A inflamação intersticial é comum e a necrose papilar renal ocorre em 20%. Raramente glomerulonefrite granulomatosa e granulomas necrosantes são observados em biópsias.
Poliangiíte microscópica
Constitucionais: febre de início insidioso, artralgia, mialgia e perda de peso.
Renais: manifestação bem prevalente na PAM (80% dos casos) indo desde achados assintomáticos até lesão renal terminal. A principal característica renal do PAM é a glomerulonefrite rapidamente progressiva (GNRP). Atribuíveis à glomerulonefrite, as características clínicas mais comuns do envolvimento renal são hematúria microscópica, proteinúria e cilindros granulares ou hemáticos na urina.
Pulmonar: o envolvimento pulmonar pode ser observado em 25 a 55% dos casos. A característica pulmonar mais comum é a hemorragia alveolar difusa; entretanto, alguns pacientes podem desenvolver fibrose intersticial crônica. As manifestações comuns de uma hemorragia alveolar incluem hemoptise, tosse, dispneia e dor torácica.
Cutâneo: lesões cutâneas são observadas em 30 a 60% dos pacientes com PAM e são a manifestação inicial em 15 a 30% dos pacientes. Púrpura palpável, livedo reticular, urticária, nódulos e úlceras cutâneas com necrose.
Gastrointestinal: a dor abdominal é o sintoma gastrointestinal mais frequentemente observado na PAM. Embora o sangramento gastrointestinal possa ocorrer em alguns casos, a hemorragia intensa é rara.
Sistema nervoso: neuropatia periférica é mais frequente que o envolvimento do sistema nervoso central. As características predominantes do sistema nervoso periférico incluem polineuropatia simétrica distal e mononeurite múltipla. A biópsia do nervo sural revela vasculite necrosante em até 80% dos casos. As manifestações do sistema nervoso central são variadas e podem incluir hemorragia cerebral, infartos cerebrais não hemorrágicos e paquimeningite.
As manifestações oculares e otorrinolaringológicas não são tão pronunciadas na PAM, porém pode acontecer.
Na histologia das biópsias: se caracterizam por vasculite necrosante de pequenos vasos (arteríolas, capilares, vênulas). Os achados pulmonares na PAM são mais comumente uma forma de capilarite difusa (distinguindo-a da GPA, que apresenta caracteristicamente lesões granulomatosas). A biópsia de pele revela vasculite leucocitoclástica aguda ou crônica com infiltrado neutrofílico nos vasos de calibre estreito da derme superficial. A biópsia renal mais comumente varia de glomerulonefrite necrosante e esclerosante difusa, focal ou segmentar, que mostra depósitos mínimos ou inexistentes de complexos imunes na microscopia óptica e de imunofluorescência (“pauci-imune”).
Critérios Classificatórios ACR 2022
– GEPA
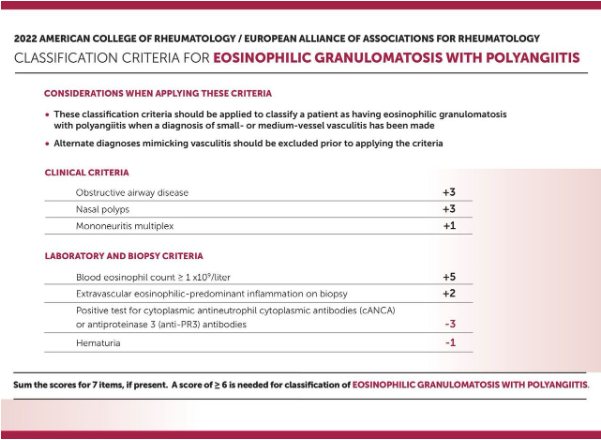
Estes critérios são validados e destinam-se ao propósito de classificação de vasculite e não são apropriados para uso no estabelecimento de um diagnóstico de vasculite. O objetivo dos critérios de classificação é diferenciar os casos de GEPA de tipos semelhantes de vasculite em ambientes de pesquisa.
Quando comparada com outras formas de VAA, no entanto, a glomerulonefrite comprovada por biópsia foi significativamente menos comum em pacientes com GEPA (4,9%) em comparação com aqueles com GPA (27,8%) ou PAM (48,5%). Da mesma forma, anti-PR3-ANCAs foram relatados em poucos pacientes com GEPA, mas são muito mais prevalentes na GPA.
Se for atingida uma pontuação cumulativa de 6 ou mais, um paciente com diagnóstico de vasculite de vasos pequenos ou médios pode ser classificado como tendo GEPA com uma sensibilidade de 85% e uma especificidade de 99%.
– GPA
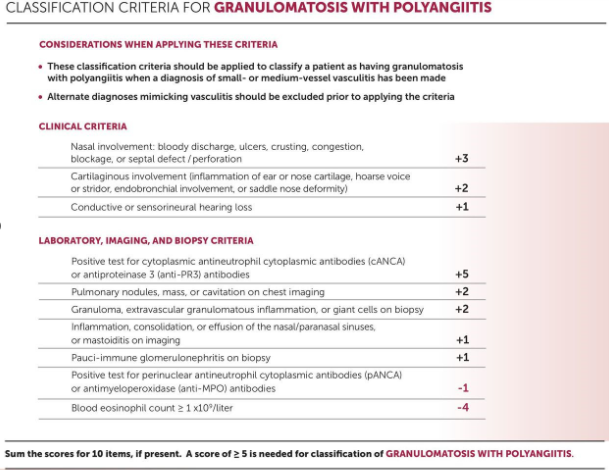
O uso de um ponto de corte ≥5 para a pontuação de risco total gerou uma sensibilidade de 92,5% e uma especificidade de 93,8%.
Estes critérios são validados e destinam-se ao propósito de classificação de vasculite e não são apropriados para uso no estabelecimento de um diagnóstico de vasculite. O objetivo dos critérios de classificação é diferenciar os casos de GPA de tipos semelhantes de vasculite em ambientes de pesquisa. Portanto, os critérios só devem ser aplicados quando tiver sido feito um diagnóstico de vasculite de pequenos ou médios vasos e todos os potenciais “simuladores de vasculite” tiverem sido excluídos.
-PAM

Uso de um ponto de corte ≥5 na pontuação de risco total, produziu uma sensibilidade de 90,8% e uma especificidade de 94,2%
Estes critérios são validados e destinam-se ao propósito de classificação de vasculite e não são apropriados para uso no estabelecimento de um diagnóstico de vasculite. O objetivo dos critérios de classificação é diferenciar os casos de PAM de tipos semelhantes de vasculite em ambientes de pesquisa. Portanto, os critérios só devem ser aplicados quando um diagnóstico de vasculite de pequenos ou médios vasos tiver sido feito e todos os potenciais “simuladores de vasculite” tiverem sido excluídos.
Métricas
O uso rotineiro de marcadores de atividade inflamatória, como proteína C reativa (PCR) e velocidade de hemossedimentação (VHS), é bastante difundido, apesar do seu valor deveras limitado no que diz respeito à atividade de doença, principalmente por reduzida especificidade
Apesar de ser um importante pilar do diagnóstico das VAA, ainda existe certa divergência na literatura em relação à elevação dos títulos ou à persistência da positividade do ANCA como marcador de atividade e preditor de recidivas.
No contexto em que há pouca concordância com relação ao uso de parâmetros sorológicos para avaliação da atividade o uso de avaliação clínica, por ferramentas, pode ser um importante aliado no manejo individual de cada paciente bem como em estudos clínicos.
Five Factor Score (FFS)
É usado para avaliação prognóstica da GEPA, prevê o risco de mortalidade. Considera manifestações ao diagnóstico como insuficiência renal (creatinina sérica >1,58 mg/dl), proteinúria >1 g por dia, cardiomiopatia, envolvimento gastrointestinal e envolvimento do sistema nervoso central. É importante ressaltar, porém, que este índice não inclui hemorragia alveolar e/ou mononeurite múltipla, condições que podem ter um impacto grave na função em pacientes com GEPA.
Birmingham Vasculitis Activity Score (BVAS)
É considerado a ferramenta validada de maior impacto e aplicabilidade prática no acompanhamento de pacientes com vasculite ANCA associada. Trata-se de um painel intuitivo e de fácil utilização após a familiarização com os itens descritos, os quais se baseiam em situações clínicas tipicamente associadas à atividade da doença de base. O objetivo do escore é o de identificar indícios de doença ativa. Constam 56 itens, que dizem respeito ao envolvimento de nove órgãos/ sistemas, sendo atribuídas pontuações com peso, determinadas de forma a dar relevância a manifestações com maior gravidade. A pontuação deve ser dada quando fenômeno descrito for secundário à vasculite ativa (tendo excluído mimetizadores, como infecção por exemplo). A pontuação vai de 0 a 63 para situações de atividade de doença com a aparição de novos itens ou a exacerbação de itens do BVAS3 que estavam presentes. Para itens persistentes, a pontuação varia de 0 a 33.
No primeiro encontro com o paciente, todas as variáveis atribuídas à atividade devem ser pontuadas, independentemente do tempo decorrido desde o início da manifestação até a primeira avaliação. Em consultas subsequentes, somente serão levados em conta os itens considerados novos ou quando há clara piora de uma determinada variável nas últimas quatro semanas.
O alvo final dos diversos regimes terapêuticos possíveis nas VAA é sempre o de remissão. Um trunfo interessante do escore é estabelecer uma definição precisa para tal, que se reflete num escore de zero. Nos casos em que a doença permanece determinando alterações representadas no BVAS, porém sem suscitar a necessidade de mudança na terapia imunossupressora, apenas a manutenção da terapia vigente, fica estabelecida a definição de doença persistente, ou seja, quando não há aparecimento de novas manifestações ou exacerbação de manifestação pré-existente
ANCA-Associated Vasculitis Patient-Reported Outcomes Questionnaire (AAV-PRO)
Incorporado sintomas importantes para o paciente (mesmo que esses sintomas não causem risco de vida).
Questionário que engloba seis diferentes domínios através de 29 itens e é respondido pelo próprio paciente, sem interferência do médico.
Os domínios englobam a gravidade dos sintomas órgão-específicos, gravidade dos sintomas sistêmicos, efeitos colaterais ao tratamento, impacto social e emocional, preocupações sobre o futuro e funcionalidade física.
Vasculitis Damage Index
É um instrumento validado para registrar danos em VAA e fornece definições que ajudam a distinguir danos de doença ativa.
Aborda 10 órgãos e sistemas, além de um 11º domínio (outros danos). O escore varia de 0 até 64. Deve ser pontuado como dano apenas manifestações que aparecerem após o início da vasculite e que persistam por mais de três meses. O dano é sempre cumulativo e o paciente não apresenta redução na pontuação no escore de dano. Essa pontuação pode manter-se estável ou aumentar. Apesar da resolução das queixas, o paciente deverá manter a mesma pontuação. O VDI maior do que 4 aumenta a mortalidade em dois anos.
Diagnóstico
Biópsia além de apoiar um diagnóstico clínico, as biópsias (particularmente do rim) podem ser úteis para distinguir a doença ativa dos danos como causa do declínio clínico. As barreiras às biópsias podem incluir dificuldade de acesso ao tecido (por exemplo, massa retro-orbital na GPA), risco injustificado do procedimento (por exemplo, pacientes que estão em terapia anticoagulante) e baixo rendimento previsto (por exemplo, as sensibilidades diagnósticas das vias aéreas superiores e biópsias transbrônquicas são apenas 30% e 12%, respectivamente).
Quando a obtenção ou interpretação de uma biópsia é desafiadora, marcadores substitutos podem apoiar um diagnóstico clínico de VAA baseado em uma apresentação clínica típica e sorologia positiva para proteinase 3 (PR3)-ANCA ou mieloperoxidase (MPO)-ANCA.
Testes adicionais para anticorpos contra a membrana basal glomerular (anti-MBG) são aconselháveis no contexto da síndrome pulmonar-renal, uma vez que os pacientes com sobreposição anti-MBG/VAA apresentam menor sobrevida renal
Esses parâmetros substitutos podem ser clínicos (como mononeurite múltipla confirmada por estudos eletrofisiológicos), dados laboratoriais (como cilindros de glóbulos vermelhos na urina sugestivos de glomerulonefrite) ou achados em exames de imagem.
A TC do tórax é mais sensível do que as radiografias convencionais e ajuda a distinguir as manifestações da doença da VAA de infecções e outras comorbidades, e a detectar doença pulmonar intersticial em pacientes com PAM.
A ressonância magnética pode detectar lesões do sistema nervoso central, paquimeningite, lesões retro-orbitais ou inflamação subglótica na GPA ou doença cardíaca na GEPA.
A endoscopia contribui para o tratamento de certas manifestações específicas de órgãos, como estenose subglótica ou brônquica, ou vasculite do trato gastrointestinal.
O lavado broncoalveolar contribui para a avaliação de infiltrações pulmonares, particularmente hemorragia alveolar ou alveolite eosinofílica, e para análise microbiológica do trato respiratório inferior.
GEPA
O diagnóstico deve ser considerado em pacientes com asma, rinossinusite crônica e eosinofilia que desenvolvem envolvimento de órgãos-alvo, particularmente neuropatia periférica, infiltrados pulmonares, cardiomiopatia ou outras complicações (por exemplo, envolvimento cutâneo, gastrointestinal ou renal).
Deve basear-se em características clínicas altamente sugestivas, evidências objetivas de vasculite (por exemplo, a partir da histologia) e ANCA.
Deve ser descartar outras doenças eosinofílicas e vasculíticas e investigado as principais complicações da doença.
Avaliação:
Geral: hemograma, urina 1, proteunúria, proteina c reativa, sangue oculto nas fezes (avaliação intestinal)
Infecciosa: escarro pulmonar nos casos duvidos, cultura de fezes, sorologias.
Cardiaco: ecocardiograma, troponina, pró BNP, ressonância cardíaca.
Nariz, ouvido e garganta: avaliação de um otorrinolaringologista, tomografia de face, audiometria.
Pulmonar: prova de função pulmonar e imagem pulmonar (radiografia ou tomografia).
Neurológica: eletroneuromiografia.
Anormalidade renal: biópsia renal. Os tecidos renais geralmente apresentam glomerulonefrite necrosante crescente que pode ser acompanhada por infiltrados eosinofílicos, alterações granulomatosas e vasculite necrosante (rica em eosinófilos) de arteríolas e artérias. Também podem ocorrer apresentações renais atípicas com outras glomerulopatias, como nefropatia membranosa, principalmente em pacientes ANCA-negativos.
Cutâneo: biópsia, vasculite necrosante de pequenas artérias que pode ser acompanhada por granulomas extravasculares. Os eosinófilos teciduais podem estar distribuídos em padrão vascular, perivascular ou dérmico intersticial.
Diferencias que pode levar eosinofilia: são numerosos e têm diferentes etiologias, incluindo formas alérgicas, condições hematológicas (por exemplo, síndromes hipereosinofílicas linfocíticas e mieloproliferativas, esta última caracterizada pela fusão dos genes FIP1L1 ), infecções parasitárias e distúrbios de hipersensibilidade, como aspergilose broncopulmonar alérgica. Outras condições que apenas ocasionalmente se apresentam com eosinofilia, mas que podem ter características sobrepostas à EGPA (tais como infecção por HIV ou doença relacionada com IgG4) também devem ser consideradas.
GPA
Não existem critérios diagnósticos para GPA. O diagnóstico é baseado em uma combinação de manifestações clínicas, sorologia positiva para ANCA e evidência histológica de vasculite necrosante, glomerulonefrite necrosante ou inflamação granulomatosa de uma biópsia de órgão relevante, como pele, pulmão ou rim.
Deve-se enfatizar que a sorologia positiva para ANCA não é essencial para o diagnóstico de GPA.
A positividade do c-ANCA (proteinase 3) é altamente específicos para GPA, especialmente na fase ativa da doença.
A maioria das amostras de biópsia na região da cabeça e pescoço apresenta achados inespecíficos. A tríade clássica de vasculite, necrose e inflamação granulomatosa pode ser observada em até 16% dos casos de GPA.
Diagnósticos diferenciais: vasculite reumatoide, pioderma gangrenoso, linfoma, tuberculose, sarcoidose e infecção fúngica profunda, síndrome de Ménière.
PAM
Não existe critérios diagnósticos, apenas critérios de classificação.
O diagnóstico se baseia no conjunto de achados.
A avaliação de um paciente com suspeita de poliangiíte microscópica envolve uma avaliação clínica, radiológica, histopatológica e laboratorial.
Nos exames podemos encontrar:
Hemograma: pode apresentar leucocitose e anemia.
Elevação de provas inflamatórias
Elvação de creatinina e ureia. Sedimentos urinários anormais.
ANCA positivo em até 80% dos casos, principalmente P-ANCA. Recomendado a dosagem MPO-ANCA (PR3-ANCA pode estar presente em até 30% dos casos).
Complemento normal.
Hemoculturas.
Tomografia de tórax: o achado mais comum (94%) na tomografia computadorizada é a atenuação em vidro fosco, que corresponde à hemorragia alveolar, inflamação crônica intersticial dos septos alveolares e capilarite.
ENMG se sinais de alteração neurológica periférica. Endoscopia digestiva alta e colonoscopia se sintomas gástricos/intestinais.
Avaliação histopatológica: deve ser feito quando possível (biópsia de pele, renal e pulmonar) para procurar evidências de vasculite e depósitos imunológicos.
Diagnósticos diferenciais: endocardite, infecções fúngicas disseminadas, linfoma, uso de cocaína (levamisol), amiloidose, sarcoidose, outras vasculites ANCA, sindrome anticorpo antimembrana basal e crioglobulinemia.
ANCA
No início da década de 1990, os critérios de classificação e a nomenclatura para as vasculites de pequenos vasos foram atribuídos pelo American College of Rheumatology e pela Chapel Hill Consensus Conference (CHCC). Esses critérios foram baseados em manifestações clínicas e características patológicas marcantes de amostras de biópsia de tecido, mas não incorporaram testes ANCA. As vasculites de pequenos vasos foram originalmente consideradas apenas associadas ao ANCA, mas estudos subsequentes em modelos animais mostraram que os ANCAs também têm potencial patogênico.
O fato de serem necessárias diferentes abordagens para demonstrar o potencial patogénico dos MPO-ANCAs e PR3-ANCAs nestes estudos aumentou a consciência de que em vez de distinguir entre pacientes com GPA, PAM e GEPA, diferenciar entre pacientes com MPO-ANCAs ou PR3-ANCAs pode ser mais relevante clinicamente.
O potencial papel patogênico combinado desses autoanticorpos e o bom desempenho dos testes de ANCA formaram a base para a incorporação de ANCAs nos critérios de nomenclatura atualizados de 2012 Chapel Hill Consensus Conference (CHCC).
A pesquisa de ANCAs associados à vasculite é realizada por um imunofluorescência indireta (IFT). Existem três principais padrões.
A primeira, C-ANCA, é uma fluorescência granular citoplasmática difusa que é mais proeminente centralmente; é observado em 90% dos pacientes com GPA generalizada e ativa.
O segundo, P-ANCA, é um padrão de coloração perinuclear de neutrófilos, muitas vezes contendo extensão nuclear; é encontrado em pacientes com PAM e GEPA.
O terceiro, denominado padrão atípico, denominado A-ANCA, é raro e combina coloração citoplasmática e perinuclear; A-ANCA pode ocorrer em associação com exposição a medicamentos, doença inflamatória intestinal ou artrite reumatóide, mais frequentemente na ausência de vasculite.
Os principais antígenos alvo reconhecidos pelos ANCAs no AAV são a proteinase 3 (PR3) e a MPO.
PR3-ANCA geralmente causa um padrão C-ANCA e está associado principalmente ao GPA.
MPO-ANCA estão associados a um padrão P-ANCA e são observados predominantemente em pacientes com PAM.
O desempenho dos ensaios imunoenzimáticos (ELISAs) melhorou e foram introduzidas tecnologias novas, sensíveis e automatizadas, como imunoensaios fluoroenzimáticos, ensaios de quimioluminescência e imunoensaios de fluxo multiplexado.
Para esse método é usado amostras com material de referência de soro humano.
Além disso, a configuração do ensaio (apresentação do antígeno) avançou com o desenvolvimento de ensaios de segunda geração (baseados em captura) e de terceira geração (baseados em âncora).
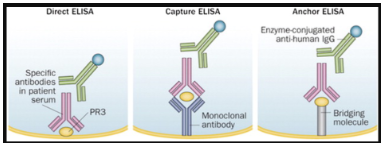
A disponibilidade de imunoensaios antígeno-específicos confiáveis levantou dúvidas se a estratégia diagnóstica em dois estágios, iniciando pela IFI é a melhor abordagem (IFI positivo e depois ELISA). Sendo assim as recomendações sobre ANCA de 2017, recomendam o uso de imunoensaios de alta qualidade (ELISA) como o primeiro método de triagem preferido para GPA e PAM. A IFI não é mais considerada adequada como primeiro teste de triagem e acrescenta poucos benefícios adicionais aos ensaios antígeno-específicos no diagnóstico de VAA quando a probabilidade pré-teste para a doença é alta.
Em pacientes com alto grau de suspeita clínica e resultados negativos do teste ANCA, ou pacientes com baixo grau de positividade (podendo ser um falso positivo), a testagem por outro método pode ser útil para aumentar a sensibilidade (ou um segundo ELISA ou IFI)
O ANCA é detectável na maioria dos pacientes com GPA e PAM recentemente diagnosticados e contribui para o diagnóstico. Embora o ANCA seja uma ferramenta sensível e específica para apoiar o diagnóstico de VAA, o diagnóstico não deve ser feito apenas com base na sorologia do ANCA, pois o ANCA pode ser encontrado em outras doenças inflamatórias e infecções, ou pode ser induzido por medicamentos, assim como, um diagnóstico de vasculite associada a ANCA não pode ser excluído com base em resultados negativos de PR3-ANCA e MPO-ANCA (doença limitada ao trato respiratório ou com vasculite renal limitada podem ser ANCA negativos).
Nos teste de ELISA tradicionalmente, um único valor de corte é empregado para prever a reatividade clinicamente relevante. No entanto, muita informação é perdida quando apenas os resultados binários (positivos ou negativos) são considerados, enquanto a probabilidade de VAA aumenta com o aumento dos níveis de PR3-ANCAs e MPO-ANCAs. Uma análise detalhada do grande conjunto de dados do estudo multinacional EUVAS exemplificou e confirmou que a razão de verossimilhança para VAA aumenta com o aumento dos níveis de PR3-ANCAs e MPO-ANCAs para todos os imunoensaios incluídos no estudo.
Com prevalência de 30% ao diagnóstico na GEPA (33,0% e 30,6% para IFI e ELISA), o ANCA é menos frequente, nos quais o MPO-ANCA é o sorotipo predominante (A proporção de MPO-ANCA entre todos os pacientes com EGPA positivo para ANCA variou de 71,4% a 100%, com mediana de 93,3%).
GEPA com PR3-ANCA compartilha características clínicas com GPA. Glomerulonefrite e neuropatia ocorrem mais frequentemente em GEPA ANCA-positivos, enquanto infiltrados pulmonares e cardiomiopatia são mais frequentes em pacientes ANCA-negativos (síndromes diferentes).
A maioria dos estudos que avaliaram uma associação entre a positividade do ANCA e as características clínicas predominantes da GEPA mostraram que os pacientes ANCA-positivos tinham maior probabilidade de apresentar manifestações de vasculite, como glomerulonefrite, neuropatia periférica, hemorragia alveolar ou púrpura, e menos frequentemente apresentavam doença cardíaca ou doença granulomatosa. envolvimento pulmonar em comparação com pacientes ANCA-negativos.
Contudo, as evidências disponíveis sugerem que a vasculite em pacientes com GEPA pode ocorrer tanto na presença como na ausência de ANCA. Sendo assim, o MPO-ANCA não pode ser utilizado como substituto único da vasculite sistémica.
Em vários estudos, os pacientes com GEPA positivo para ANCA tiveram maior probabilidade de serem tratados com ciclofosfamida ou doses maiores de glicocorticoide. Os pacientes ANCA-positivos tiveram maior probabilidade de alcançar a remissão quando receberam rituximabe do que os pacientes ANCA-negativos
A positividade para ANCA em pacientes com GEPA pode sinalizar a necessidade de terapia imunossupressora mais intensiva (por exemplo, adição de ciclofosfamida ou rituximabe) porque podem estar presentes características de vasculite que geralmente não são suficientemente tratadas com glicocorticoides em monoterapia. Contudo, a positividade do ANCA por si só não indica necessidade de intensificação do tratamento.
Não foram observadas diferenças na resposta ao tratamento entre pacientes ANCA-positivos e ANCA-negativos. Anticorpos antilactoferrina foram detectados em pacientes com GEPA.
Medições seriadas de ANCA podem ter algum valor na previsão de recidivas de GPA e PAM em pacientes com vasculite renal e/ou hemorragia alveolar. Na GEPA, o monitoramento do ANCA só é útil quando o teste MPO-ANCA foi positivo no início da doença. A repetição do teste de ANCA é recomendada em pacientes com GEPA positivo para MPO-ANCA porque a persistência, aumento ou reaparecimento de ANCA pode justificar uma avaliação clínica mais frequente.
PR3-ANCA foi detectado em 84%–85% dos pacientes com GPA e 2%–27% dos pacientes com PAM, enquanto MPO-ANCA foi encontrado em 16% dos pacientes com GPA e 75%– 97% com PAM.
20-35% dos pacientes com doença antimembrana basal glomerular (anti-GBM) podem ter ANCA positivo.
O P-ANCA é encontrado não apenas em pacientes com MPO-ANCA, mas também em pacientes com anticorpos contra lactoferrina, elastase, catepsina G, lisozima e outros antígenos.
Pacientes com doenças gastrointestinais: além das vasculites, os ANCAs são encontrados em pacientes com distúrbios gastrointestinais, como doença inflamatória intestinal, colangite esclerosante primária e doenças inflamatórias do fígado (como hepatite autoimune, cirrose biliar primária e hepatite viral crônica). Estas doenças estão associadas a um padrão P-ANCA ligeiramente aumentado, frequentemente referido como P-ANCA ou ANCA atípico.
A combinação de P-ANCA com medições de anticorpos anti- Saccharomyces cerevisiae (ASCA) pode melhorar a utilidade clínica deste marcador. ASCAs são encontrados em 40–60% dos pacientes com doença de Crohn, 4–14% dos pacientes com colite ulcerosa e <5% dos controles. A combinação de um resultado de teste ASCA positivo e P-ANCA negativo está associada à doença de Crohn, enquanto a combinação de um resultado de teste ASCA negativo e P-ANCA positivo está associada à colite ulcerosa. Estudos também relataram que imunoensaios sensíveis podem detectar PR3-ANCAs em pacientes com colite ulcerativa e colangite esclerosante primária. A coexistência de VAA com DII foi descrita em alguns pacientes, mas é rara.
ANCAs também foram relatados em doenças sistêmicas, como artrite reumatóide e lúpus eritematoso sistêmico, além de neoplasias como linfoma não-Hodgkin e mielodisplasia.
A endocardite infecciosa pode mimetizar a glomerulonefrite associada ao ANCA e os pacientes com endocardite infecciosa podem desenvolver ANCAs.
A cocaína adulterada com levamisol (a dupla positividade para MPO-ANCAs e PR3-ANCAs é uma característica desta doença, porém sendo mais frequente o MPO-ANCA) e drogas como hidralazina, propiltiouracil e minociclina podem causar formas secundárias de VAA. A maioria dos pacientes com VAA induzida por medicamentos apresenta MPO-ANCA, que pode ser encontrado em combinação com anticorpos para outras proteínas citoplasmáticas de neutrófilos ( como elastase de neutrófilos humanos, lactoferrina, catepsina G e proteína aumentadora de permeabilidade bactericida) e FAN.
A dosagem e a intensidade do tratamento não deve ser baseado apenas na titulação do ANCA. Aumentos nos títulos/níveis de ANCA são apenas modestamente informativos como um indicador da atividade da doença e não são preditores confiáveis de exacerbações da doença para pacientes individuais. A persistência da positividade para ANCA não indica necessariamente que seja necessária terapia imunossupressora continuada. Em vez disso, as decisões de tratamento devem basear-se nos sintomas clínicos do paciente em conjunto com estudos diagnósticos (por exemplo, resultados laboratoriais, de imagem e de biópsia).
A fim de garantir o uso adequado do teste de anticorpo anticitoplasma de neutrófilos (ANCA) para apoiar o diagnóstico de vasculite associada a ANCA (AAV), o ANCA deve ser solicitado para pacientes com as seguintes indicações clínicas.
- Glomerulonefrite, especialmente glomerulonefrite rapidamente progressiva
- Hemorragia pulmonar, especialmente síndrome renal pulmonar
- Vasculite cutânea com características sistêmicas
- Múltiplos nódulos pulmonares
- Doença destrutiva crônica das vias aéreas superiores
- Sinusite ou otite de longa duração
- Estenoses traqueais subglóticas
- Mononeurite múltipla ou outra neuropatia periférica
- Massa retro-orbital
- Esclerite
O Teste ANCA deve ser solicitado para apoiar um diagnóstico de GEPA, se o paciente apresentar asma ou rinossinusite e eosinofilia sanguínea associado com uma ou mais das seguintes características:
- Febre, perda de peso, artralgia, mialgia com sinais laboratoriais de inflamação
- Infiltrados pulmonares
- Neuropatia sensorial ou motora periférica
- Doença cardíaca inexplicável (arritmias ou insuficiência cardíaca)
- Alteração de função renal ou proteinúria associada com hematúria.
- Hemorragia alveolar
- Dor abdominal
- Púrpura ou erupções cutâneas
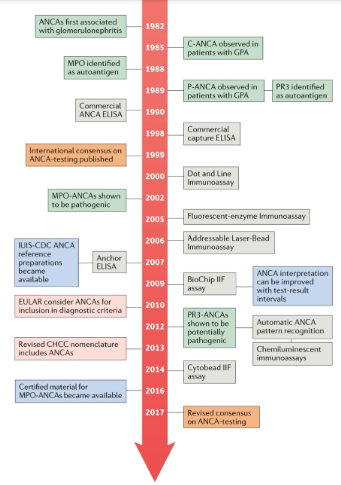

Bossuyt, X. et al. (2017) Revised 2017 international consensus on testing of ANCAs in granulomatosis with polyangiitis and microscopic polyangiitis.Nat. Rev. Rheumatol. doi:10.1038/nrrheum.2017.140
Tratamento
Os pacientes com VAA devem receber os melhores cuidados, baseados na tomada de decisão compartilhada (médico paciente), considerando a eficácia, a segurança e os custos.
Educação do paciente sobre a doença, sobre sintomas de alerta (tanto da doença ativa como complicações da imunossupressão).
Esses pacientes devem ser seguidos de perto pelo médico, tanto para avaliar resposta ao tratamento como as complicações (da doença e do tratamento). Por exemplo: infecção, neoplasia de bexiga (uso de ciclofosfamida), diabetes mellitus e aumento da pressão arterial induzidas pelo corticoide, risco de osteoporose, etc.
Para pacientes com envolvimento nasossinusal na GPA, lavagens nasais e terapias nasais tópicas (antibióticos, lubrificantes e GCs) podem ser benéficas.
Para pacientes com GPA em remissão que apresentam defeitos do septo nasal e/ou colapso da ponte nasal, cirurgia reconstrutiva, se desejado pelo paciente pode ser realizada.
Para pacientes com GPA e tecido subglótico e/ou endobrônquico ativamente inflamado com estenose, recomendado o tratamento com terapia imunossupressora em vez de dilatação cirúrgica.
Para pacientes com GPA e lesões de massa (por exemplo, pseudotumor orbital ou massas das glândulas parótidas, cérebro ou pulmões), recomendado o tratamento com terapia imunossupressora em vez da remoção cirúrgica da lesão.
Para pacientes com GPA/PAM em remissão e doença renal crônica em estágio 5, considerar transplante renal.
O uso de inibidores de leucotrienos não é contraindicado para pacientes com GEPA com asma ativa e/ou doença nasossinusal. Se o paciente já usa, pode manter o uso.
Indução
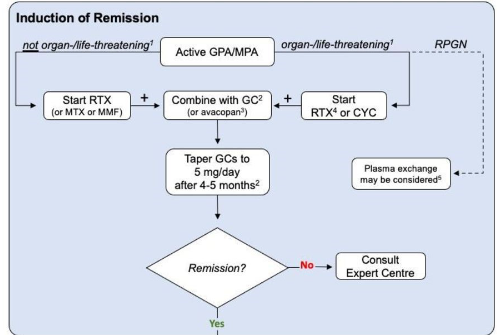
Para indução da remissão em pacientes com GPA ou PAM de início recente ou recidivante com doença que ameaça órgãos ou com risco de vida, é recomentado o tratamento com uma combinação de glicocorticoide e rituximabe ou ciclofosfamida (CYC; 2mg/kg/dia oral por 3-6 meses ou 15mg/kg/dia EV a cada 15 dias por 3 doses e após 15mg/kg a cada 3 semanas por pelo menos 3 doses).
O Rituximabe (RTX) é preferido na doença recidivante.
No maior estudo comparando RTX e CYC para indução de remissão, as taxas de remissão aos 6 e 12 meses em pacientes com recidiva foram maiores para RTX.
Uma meta-análise que incluiu estudos retrospectivos descobriu que os resultados de eficácia e segurança não diferem entre o protocolo RTX usado no estudo RAVE (375 mg/m 2 por semana durante 4 semanas), e o protocolo de duas doses (1 g nas semanas 0 e 2) aprovado para artrite reumatóide.
O recente estudo PEXIVAS incluiu pacientes com doença renal grave e/ou hemorragia alveolar difusa (HAD) tratados com RTX e os resultados parecem não diferir em comparação com o CYC, mas o estudo não foi suficientemente poderoso para demonstrar a não inferioridade do RTX sobre o CYC em este subgrupo (talvez a CYC seja mais indicada nesses grupo de pacientes).
Dada a provável menor eficácia a longo prazo em pacientes com VAA positivo para PR3-ANCA, a falta de superioridade em segurança e a falta de aprovação formal para uso em VAA, não há evidências suficientes para apoiar o uso rotineiro de MMF como um tratamento de primeira escolha para GPA ou PAM de início recente em vez de RTX ou CYC. O uso do micofenolato mofetil (3000mg/dia divido em 2x) para indução da remissão deve ser limitado a situações em que o RTX e o CYC não são tolerados ou são contraindicados.
Para os casos sem ameaça aos órgãos ou à vida, recomenda-se o tratamento com uma combinação de glicocorticoide e RTX. O metotrexato ou micofenolato podem ser considerados alternativas ao RTX. O estudo RAVE e ensaios recentes usando RTX para terapia de indução incluíram esses pacientes, e os resultados de eficácia e segurança não foram inferiores em comparação com aqueles que tinham doença mais grave no início do estudo.
O estudo NORAM comparando CYC oral versus MTX oral em GPA de início recente mostrou que 50% dos pacientes no grupo MTX não atingiu a remissão ou apresentou recaída no mês 12 (isso em doses mais altas de corticoide do que o indicado atualmente)
Estudo RITAZAREM mostrou que 66 dos 69 pacientes com GPA ou PAM sem manifestação de risco de órgãos que foram tratados com RTX estavam em remissão no mês 4, apesar do uso de um regime de GC em dose mais baixa com uma dose inicial de 30 mg de prednisolona por dia.
Assim sendo, o uso de RTX em vez de metotrexato ou micofenolato deve ser considerado em pacientes com GPA e PAM, mesmo sem manifestações que ameacem os órgãos, uma vez que regimes de indução e remissão baseados em RTX estão associados a taxas mais altas de remissão sustentada e menor exposição a glicocorticoide.
O glicocorticoide (GC) faz parte do tratamento de indução, é indicado uma dose de inicial de 50-75 mg equivalente de prednisolona/dia com redução gradual para chegar em 5mg/dia em 4 à 5 meses. O estudo PEXIVAS comparou dois regimes de redução gradual de glicorticoide em pacientes com GPA e PAM e doença ativa com risco de vida ou com risco de órgãos. O regime de dose reduzida resultou em uma redução de 40% na exposição oral ao GC nos primeiros 6 meses e não foi inferior. O estudo RITAZAREM usou a dose 0,5mg/kg e 1 mg/kg/dia e quando os pacientes foram estratificados para recidiva “maior” ou “menor”, não foram observadas diferenças na eficácia para nenhum dos subgrupos de gravidade entre as duas doses.
Doses iniciais mais baixas de GC de 0,5 mg/kg/dia podem ser consideradas individualmente em pacientes selecionados sem doença com risco de vida ou com risco de órgãos.
A administração de pulsos intravenosos de metilprednisolona (MP) em doses de 1.000 a 3.000 mg tem sido usada em protocolos de indução. A melhor evidência disponível é derivada de comparação indireta entre diferentes ensaios. Estudos observacionais não relataram nenhum benefício de eficácia, mas aumentaram as taxas de infecções com o uso de doses iniciais mais altas de GC, incluindo pulsos de MP. Tendo em conta esta limitação, e com base nas evidências do ensaio PEXIVAS, a pulsoterapia com MP deve ser limitada ao tratamento de manifestações graves que ameaçam os órgãos, particularmente envolvimento renal ativo com uma taxa de filtração glomerular estimada documentada (TFGe) de <50 mL/ min/1,73/m 2 ou na hemorragia alveolar difusa.
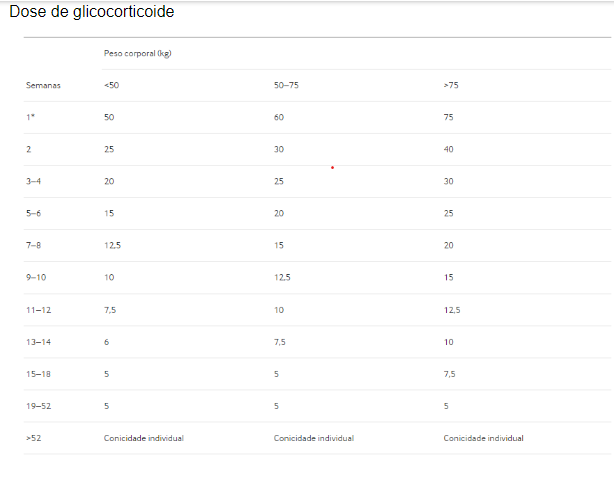
Durante a fase de indução, pode se considerar associar Avacopan (uma molécula que bloqueia o receptor C5a do complemento, na dose de 30 mg duas vezes por dia por 1 ano) ao RTX ou CYC com o objetivo de reduzir a dose do GC, baseada nos resultados do ADVOCATE RCT.
A plasmaférese pode ser considerada para pacientes com PAM ou GPA e com disfunção renal importante ( Cr sérica maior que 3,39 mg/dl). O estudo PEXIVAS foi realizado para avaliar a eficácia do plasmaférese como adjuvante da terapêutica de indução padrão em pacientes com PAM ou GPA recém-diagnosticados ou recidivantes, com PR3 ou MPO-ANCA positivo que apresentavam envolvimento renal ativo com TFGe <50 mL/min/1,73 m 2 ou hemorragia alveolar difusa. Nenhuma diferença para o desfecho primário composto de morte por qualquer causa ou DRC foi encontrada entre pacientes. Como a creatinina sérica basal prediz o risco de doença renal terminal, subgrupos com base na creatinina basal com baixo risco (≤2,2mg/dl), risco baixo a moderado (>2,2-3,39mg/dl), risco moderado a alto (>3,39–5,65 mg/dl ) e alto risco (>5,65 µmol/L) foram analisados. Embora tenha sido observada pouca redução do risco absoluto de DRC após o uso de plasmaferese nos grupos de baixo e baixo-moderado, uma redução absoluta de 4,6% de DRC em 12 meses foi estimada para o grupo moderado-alto e 16,0% para o grupo de alto risco. Esse impacto da diferença diminui após 3 anos, e além disso, em média, tratar 14 pacientes com plasmaférese resultará em 1 infecção grave.
A plasmaférese é recomendado para pacientes com VAA também positivos para anticorpos anti-membrana basal.
Para aqueles pacientes com GPA ou PAM e doença refratária a terapia padrão inicial realize uma reavaliação completa do estado da doença e das comorbidades e considerar opções para uso de tratamento adicional ou diferente. Mudança de CYC para RTX ou vice-versa. Aumentar a dose de GC por algum tempo pode ser uma estratégia razoável, principalmente se persistirem apenas sintomas leves. A combinação de RTX e CYC é usada em pacientes com doença refratária com risco de órgãos ou com risco de vida por muitos centros, mas faltam dados sobre esta abordagem. A adição de imunoglobulinas intravenosas (por exemplo, 2 g/kg) pode ser uma opção para manifestações persistentes da doença, particularmente em pacientes com risco aumentado de infecção.
Manutenção
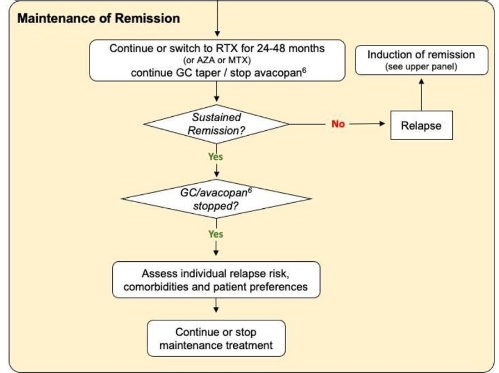
Após a indução com RTX ou CYC, a manutenção é feita com rituximabe. Azatioprina (2mg/kg/dia) e metotrexato (até 25mg/semana) podem ser opções alternativas.
MAINRITSAN: dose 1g e após 500mg a cada 6 meses por 3 vezes (duração total de 2 anos). Mostrou menos recidiva em comparação a azatioprina.
RITAZAREM: 1g a cada 4 meses por 5 doses, mostrou maior eficacia do RTX em comparação com azatioprina.
Diante dessas diferentes doses, a recomendação EULAR favorece o uso do regime de manutenção de RTX de 500 mg a cada 6 meses. A dose mais elevada de 1 g ou um intervalo de dosagem mais curto de 4 meses ou ambos podem ser considerados para pacientes que recidivam com o regime de 500 mg a cada 6 meses.
AZA e MTX são agentes de manutenção igualmente eficazes no VAA e podem ser usados se o RTX for contraindicado.
O micofenolato mofetil foi associado a uma maior taxa de recidiva em comparação com o AZA.
Em pacientes com GPA, a leflunomida pode ser considerada em pacientes com intolerância a todos os medicamentos acima mencionados.
Duas metanálises recentes revelaram que sulfametoxazol-trimetoprim (T/S) não reduz o risco de recaída em pacientes com GPA.
O tempo da terapia de manutenção recomendado é de pelo menos 24 a 48 meses após a indução. Uma duração mais longa pode ser considerado nos pacientes com recidivas ou naqueles com risco de recidiva aumentado.
No estudo REMAIN comparou AZA por 24×48 meses (os pacientes de 48 meses tiveram menos recidiva). Na fase aberta do MAINRITSAN-3 os pacientes que continuaram recebendo rituximabe após 18 meses permaneceram livres de recidiva.
O tipo de doença clínica (GPA vs MPA, risco maior na GPA), o sorotipo ANCA (PR3-ANCA vs MPO-ANCA, risco maior para PR3) e o status ANCA (positivo vs negativo, persistencia do ANCA positivo ou voltar a ser positivo foi associado a risco de recidiva) foram todos associados ao risco de recaída. Hematuria persistente, uso de MTX/micofenolato ao inves de outras terapias também foram associadas a um risco mair de recidiva
Recomendações EULAR GPA/PAM
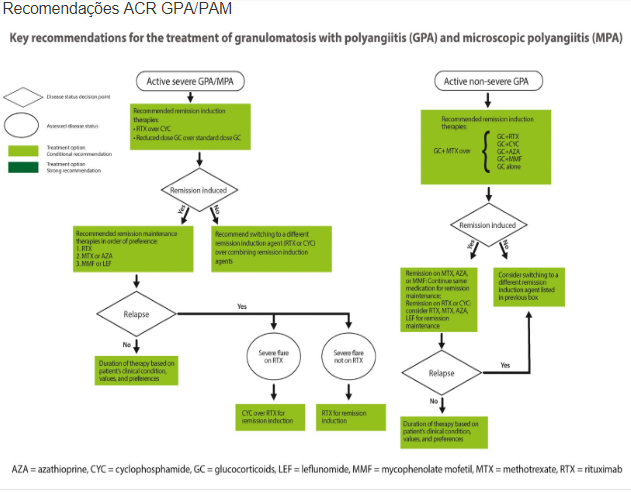
Indução na GEPA
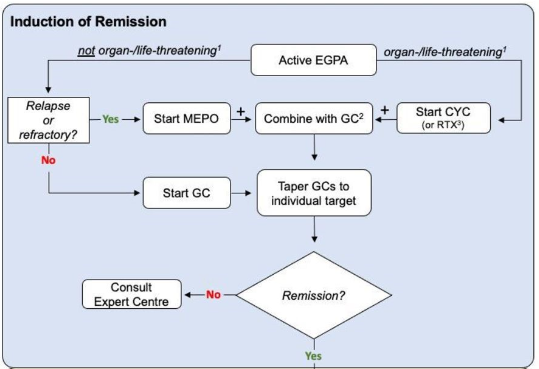
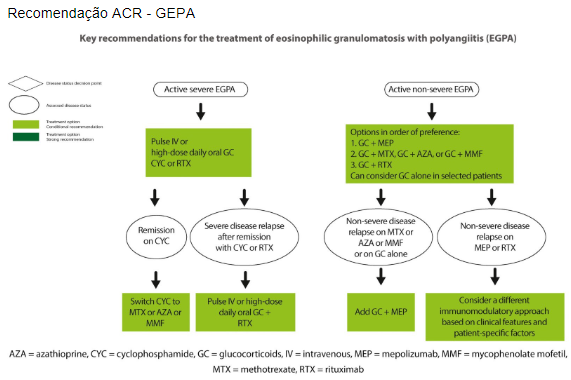
Início recente ou recidivante com manifestações que ameaçam órgãos ou ameaçam a vida, recomendamos o tratamento com uma combinação de GCs em altas doses e CYC. Rituximabe pode ser uma alternativa no lugar da CYC.
Pode ser considerado manifestações que ameaçam a vida como: insuficiência renal (creatinina sérica >1,58 mg/dl), proteinúria >1 g por dia, cardiomiopatia, envolvimento gastrointestinal e envolvimento do sistema nervoso central e periférico e mais raramente hemorragia alveolar ou algumas formas de envolvimento ocular (como oclusão da artéria ou veia central da retina, neuropatia óptica isquêmica, miosite orbital e vasculite, infartos ou edema da retina).
Recomendado que o tratamento seja alterado para uma terapia de manutenção menos intensiva após seis pulsos de CYC se a remissão for alcançada, e a dose de GC seja reduzida até então para aproximadamente 7,5 mg por dia.
Estudo que avaliou rituximabe e ciclofosfamida com o REOVAS, monstrando que o rituximabe pode ser uma opção nos casos que não é possível usar CYC.
GEPA refratário é definido como atividade da doença inalterada ou aumentada após 4 semanas de terapia apropriada de indução de remissão. A persistência ou agravamento das manifestações sistêmicas deve ser diferenciada das manifestações respiratórias. Nesses casos é importante o diagnóstico primário ser reavaliado e as avaliar se as manifestações refratárias podem ser atribuíveis a outras etiologias, como infecções ou malignidades. Avaliar a adesão do paciente e diferenciar entre lesão ativa ou dano irreversível. Nos pacientes cuja doença não responde a essas abordagens, diferentes opções terapêuticas podem ser consideradas, incluindo, plasmaférese e terapia com imunoglobulina intravenosa.
Para pacientes com GEPA sistêmica refratária, apesar do tratamento de indução de remissão com altas doses de glicocorticóides mais ciclofosfamida, o uso de rituximabe é recomendado, enquanto a ciclofosfamida deve ser usada para pacientes nos quais a indução de remissão com rituximabe falha.
Para os pacientes sem manifestações que ameacem a vida é recomendado o tratamento com corticoide (alcanção a remissão em 90% das vezes, apesar de terem recidivas frequentes quando retira a medicação). Para esse grupo de paciente, não foi encontrado benefício claro da associação de azatioprina ou rituximabe para redução de recidiva/complicações. O ACR já coloca sua preferência pela associação ao mepolizumabe e como opções o metotrexato, azatioprina ou micofenolato mofetil.
Para pacientes com doença recidivante (sem risco de órgãos), é recomendado o uso de mepolizumabe (inibidor da IL-5, 300 mg por via subcutânea a cada 4 semanas). Foi avaliado no estudo MIRRA.
Em pacientes para os quais o mepolizumabe não é eficaz ou não é tolerado, AZA, MTX, MMF ou RTX podem ser considerados individualmente.
Pacientes que apresentam asma ativa ou envolvimento otorrinolaringológico, a terapia tópica e/ou inalatória deve ser otimizada. A abordagem do manejo dessas manifestações da doença deve envolver especialistas como pneumologistas e otorrinolaringologistas. Embora o uso de terapias sistêmicas (como glicocorticoides e mepolizumabe) seja a base para o controle das manifestações respiratórias da GEPA, a combinação com terapias inalatórias deve ser considerada como tratamento de suporte no controle da asma.
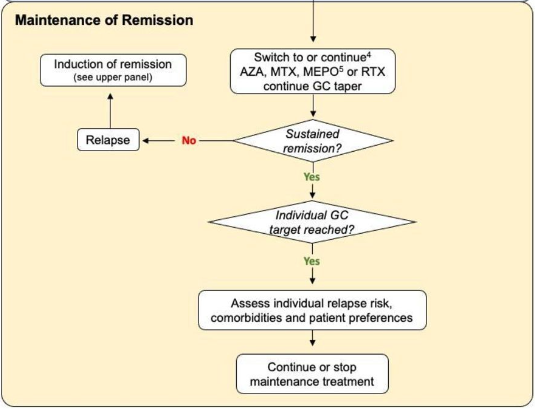
Manutenção na GEPA
Para os casos no qual foi inciado a indução com risco de órgãos, a manutenção pode ser feita com mepolizumabe, azatioprina, metotrexato ou rituximabe. O ACR coloca sua preferência para metotrexato, azatioprina ou micofenolato mofetil.
Nos casos no qual a indução não apresentava risco de órgãos, a manutenção pode ser feita com mepolizumabe.
Tendo em vista a natureza recidivante da EGPA que requer o uso prolongado de GCs na maioria dos pacientes, outros agentes para manutenção da remissão são comumente prescritos na tentativa de poupar os GC.
A remissão da GEPA é definida como a ausência de sinais ou sintomas clínicos atribuíveis à doença ativa, incluindo asma e manifestações otorrinolaringológicas. A dose diária de glicocorticóides também deve ser considerada para definição de remissão, podendo ser escolhida como ponto de corte uma dose diária máxima de 7,5 mg de prednisona.
Embora alguns parâmetros laboratoriais (por exemplo, contagem de eosinófilos ou ANCA) sejam comumente monitorados, não existem biomarcadores confiáveis para medir a atividade da doença na EGPA. A atividade da doença deve, portanto, ser avaliada no acompanhamento apenas utilizando ferramentas clínicas validadas.
Ecocardiografia e eletrocardiografia periódicas são recomendadas em todos os pacientes para detecção precoce em pacientes assintomáticos. Cuidados com infecção, vacinação, triagem de osteoporose induzida por glicorticoide e avaliação da função pulmonar.
Recomendação EULAR – GEPA
Profilaxia de Pneumocistose
Para pacientes recebendo rituximabe, ciclofosfamida ou altas doses de glicocorticoide (tratamento com GCs em uma dose ≥30 mg/dia por 4 semanas ou mais, independentemente de outros imunossupressores concomitantes) é recomendado o uso de profilaxia com bactrim (sulfametoxazol trimetoprima).
Na dose de 800/160 mg em dias alternados ou 400/80 mg diariamente.
Embora não existam dados suficientes para orientar a duração total da profilaxia com T/S, parece razoável continuar este medicamento durante a duração estimada do efeito biológico de CYC e RTX de cerca de 3 e 6 meses após a última dose ou reconstituição de células B. , respectivamente. Para pacientes tratados com GCs em combinação com imunossupressores que não sejam CYC ou RTX, a T/S pode ser interrompida assim que as doses de GC forem reduzidas gradualmente para 15 mg/dia, mas deve-se considerar fortemente a continuidade até que doses mais baixas sejam alcançadas se outros fatores de risco como doença pulmonar ou hipogamaglobulinemia estão presentes.
Para pacientes com GEPA que estão recebendo ciclofosfamida ou rituximabe, recomendado também a prescrição para profilaxia de pneumonia por Pneumocystis jirovecii.
Avaliação de hipogamaglobulinemia
A associação de terapias direcionadas às células B, como o rituximabe, com o desenvolvimento de hipogamaglobulinemia e infecção é cada vez mais reconhecida
Existem poucos dados disponíveis sobre a história natural da hipogamaglobulinemia e as indicações para terapia de reposição de imunoglobulina (IGRT) na hipogamaglobulinemia associada à terapias direcionadas às células B.
A decisão de iniciar imunglobulina deve ser tomada pelo grau de hipogamaglobulinemia, infecções (graves, persistentes, incomuns ou recorrentes), demonstração de respostas deficientes vacinais e resposta deficiente à profilaxia antibiótica.
Não existe um nível absoluto de IgG onde a imunoglobulina deva ser iniciado. A hipogamaglobulinemia é mais frequentemente assintomática e pode ser transitória neste grupo de pacientes. Sendo levado em conta a presença de infecções associadas.
A decisão de iniciar a imunoglobulina também deve considerar comorbidades individuais (por exemplo, bronquiectasia, neutropenia e co-prescrição de medicamentos imunomoduladores – particularmente corticoides).
O objetivo final da profilaxia antibiótica é que o paciente não tenha infecções bacterianas. No entanto, isto nem sempre é possível, por exemplo, em pacientes com bronquiectasias estabelecidas. Embora não existam normas atuais para definir uma resposta fraca à profilaxia antibiótica, pode-se dizer que este estado existe se houver infecções continuadas ou graves, apesar da profilaxia antibiótica relevante em curso.
Os fatores predisponentes para o desenvolvimento de hipogamaglobulinemia clinicamente significativa durante ou após receber terapias direcionadas às células B incluem um nível baixo de IgG pré-existente e terapias imunossupressoras anteriores e/ou concomitantes (rituximabe é o preditor mais importante).
Pacientes recebendo rituximabe, é recomendado a medição das concentrações séricas de imunoglobulina antes de cada ciclo de RTX para detectar imunodeficiência secundária.
Pacientes com doenças reumáticas e hipogamaglobulina sintomáticos (infecções de repetição) e assintomáticos (com IgG inferior a 3g/litro), devem ser encaminhados para acompanhamento conjunto com a imunologia.
A distinção entre imunodeficiência comum variável, que também pode estar associada à DRIMs, e hipogamaglobulinemia relacionada à terapias direcionadas às células B deve ser considerada durante a revisão de Imunologia Clínica. A verificação das imunoglobulinas basais é uma etapa importante que pode ajudar a fazer essa distinção.
Os níveis de imunoglobulina devem ser medidos antes do início da terapias direcionadas às células B e repetidos a cada 6–12 meses durante o tratamento e no mínimo um ano após a interrupção.
Na hipogamaglobulinemia relacionada à terapias direcionadas às células B, a dose inicial de imunoglobulina deve ser de 0,4 g/kg/mês. A dose da imunoglobulina deve ser ajustada de acordo com a resposta clínica, níveis mínimos de IgG, frequência de infecção e fatores individuais do paciente.
A decisão de continuar a imunoglobulina deve ser revista anualmente e baseada em parâmetros clínicos e laboratoriais. Na decisão de interromper o IGRT, as considerações individuais incluiriam a eficácia, tolerabilidade e efeitos adversos do IGRT. Foram observados os riscos potenciais da IGRT, incluindo tromboembolismo e hemólise. Avaliar por quanto tempo o imunossupressor ainda será mantido e os níveis das imunoglobulinas.
Um nível baixo de IgG não é uma contraindicação absoluta para iniciar ou continuar a terapias direcionadas às células B. A decisão deve basear-se numa análise benefício-risco individualizada. A maioria dos pacientes que desenvolvem hipogamaglobulinemia não desenvolve complicações de infecção recorrente/grave. A hipogamaglobulinemia pode ser transitória.
Não há evidências disponíveis comparando o uso de profilaxia antibiótica associado com imunoglobulina na hipogamaglobulinemia sintomática causada pela terapias direcionadas às células B, entretanto, uma tentativa inicial de profilaxia antibiótica pode ser apropriada.
Para pacientes com GPA/PAM recebendo terapia de manutenção da remissão com rituximabe que apresentam hipogamaglobulinemia (por exemplo, IgG <3 g/litro) e infecções graves recorrentes,o ACR 2021 recomenda a suplementação de imunoglobulina.
A suplementação de imunoglobulina em doses de reposição (por exemplo, 400–800 mg/kg/mês) deve ser considerada se um paciente tiver hipogamaglobulinemia e apresentar infecções recorrentes. A suplementação de imunoglobulina também pode ser considerada para pacientes com hipogamaglobulinemia sem infecções recorrentes, mas com respostas vacinais prejudicadas.
A deficiência completa de IgA é um achado comum, e frequentemente assintomático, sendo encontrada incidentalmente em indivíduos saudáveis (1 em 500–800). Não é uma contraindicação à imunoglobulina, porque as reações a níveis muito baixos de IgA são raras e podem ser tomadas precauções.
Recidivas
Para pacientes com GPA/PAM que tiveram recidiva com manifestações graves da doença e não estão recebendo rituximabe para manutenção, recomendado o tratamento com rituximabe em vez de ciclofosfamida para reindução da remissão.
Para pacientes com GPA/PAM que tiveram recidiva com manifestações graves da doença enquanto recebiam rituximabe para manutenção da remissão, ACR recomenda a mudança de rituximabe para ciclofosfamida em vez de receber rituximabe adicional para reindução da remissão. Já no EULAR recomenda que se o paciente está usando rituximabe na dose 500mg a cada 6 meses, mudar para 1g a cada 4 meses, nos casos de recidiva.
Para pacientes com GEPA que tiveram recidivas com manifestações graves da doença após indução prévia da remissão bem-sucedida com ciclofosfamida, recomendado o tratamento com rituximabe em vez de ciclofosfamida para reindução da remissão.
Para os pacientes com GEPA com recidiva e manifestações não graves enquanto recebiam metotrexato, azatioprina ou micofenolato mofetil, recomendado condicionalmente a adição de mepolizumabe em vez da mudança para outro agente.
Ao definir recidiva na GEPA, recomendado distinguir a recidiva da vasculite sistêmica (recidiva sistêmica) da exacerbação isolada da asma e das manifestações otorrinolaringológicas (recidiva respiratória). Um aumento na contagem de eosinófilos sem manifestações clínicas concomitantes não deve ser considerado uma recidiva.As recidivas sistêmicas podem ser diferenciadas em graves e não graves, apresentando-se as primeiras com manifestações incluídas na FFS ou com manifestações que ameaçam a vida ou órgãos.
Prognóstico
As principais causas de morte no primeiro ano foram infecção (48%) e vasculite ativa (19%). Altas doses de glicocorticoide contribuem para o risco de infecções.
Embora não existam dados formais disponíveis, os prognósticos das vasculites sistémicas idiopáticas, como PAM e GPA, são maus sem tratamento (mortalidade de 90% após um ano). A introdução de tratamento imunossupressor agressivo, em particular ciclofosfamida em combinação com prednisona, melhorou substancialmente o prognóstico, com sobrevida cumulativa do paciente em 1 e 5 anos de 82% e 76%, respectivamente, para pacientes com envolvimento renal.
O envolvimento cardíaco tem sido associado ao aumento da mortalidade na GEPA.
Embora o status ANCA esteja associado à recaída ensaios prospectivos sobre manutenção da remissão mostraram resultados conflitantes no status ANCA ou na contagem de células B CD19+ para prever recaídas futuras em um nível considerado insuficiente para orientar as decisões de tratamento para pacientes individuais.
Como a VAA envolve múltiplos órgãos e as recidivas são frequentes, recomenda-se uma avaliação clínica estruturada durante o acompanhamento em intervalos regulares.
Referências
- Suppiah R, Robson JC, Grayson PC , et al. 2022 American College of rheumatology/european alliance of associations for rheumatology classification criteria for microscopic polyangiitis. Ann Rheum Dis 2022;81:321–6. doi:10.1136/annrheumdis-2021-221796
- Robson JC, Grayson PC, Ponte C, et al. 2022 American College of rheumatology/european alliance of associations for rheumatology classification criteria for granulomatosis with polyangiitis. Ann Rheum Dis 2022;81:315–20. doi:10.1136/annrheumdis-2021-221795
- Grayson PC, Ponte C, Suppiah R, et al. 2022 American College of rheumatology/european alliance of associations for rheumatology classification criteria for eosinophilic granulomatosis with polyangiitis. Ann Rheum Dis 2022;81:309–14. doi:10.1136/annrheumdis-2021-221794
- Jennette JC, Falk RJ, Bacon PA , et al. 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum 2013;65:1–11. doi:10.1002/art.37715
- Rona Smith, David Jayne, Peter Merkel. A RANDOMIZED, CONTROLLED TRIAL OF RITUXIMAB VERSUS AZATHIOPRINE AFTER INDUCTION OF REMISSION WITH RITUXIMAB FOR PATIENTS WITH ANCA-ASSOCIATED VASCULITIS AND RELAPSING DISEASE, Nephrology Dialysis Transplantation, Volume 35, Issue Supplement_3, June 2020, gfaa146.LB004, https://doi.org/10.1093/ndt/gfaa146.LB004
- Guillevin L , Pagnoux C , Karras A , et al. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med 2014;371:1771–80. doi:10.1056/NEJMoa1404231
- Hellmich B, Sanchez-Alamo B, Schirmer JH, et al. EULAR recommendations for the management of ANCA-associated vasculitis: 2022 update. Annals of the Rheumatic Diseases 2024;83:30-47.
- Chung, S.A., Langford, C.A., Maz, M., Abril, A., Gorelik, M., Guyatt, G., Archer, A.M., Conn, D.L., Full, K.A., Grayson, P.C., Ibarra, M.F., Imundo, L.F., Kim, S., Merkel, P.A., Rhee, R.L., Seo, P., Stone, J.H., Sule, S., Sundel, R.P., Vitobaldi, O.I., Warner, A., Byram, K., Dua, A.B., Husainat, N., James, K.E., Kalot, M.A., Lin, Y.C., Springer, J.M., Turgunbaev, M., Villa-Forte, A., Turner, A.S. and Mustafa, R.A. (2021), 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the Management of Antineutrophil Cytoplasmic Antibody–Associated Vasculitis. Arthritis Rheumatol, 73: 1366-1383.
- Sonali Wijetilleka, David R Jayne, Chetan Mukhtyar, Aftab Ala, Philip D Bright, Hector Chinoy, Lorraine Harper, Majid A Kazmi, Sorena Kiani-Alikhan, Charles K Li, Siraj A Misbah, Louise Oni, Fiona E Price-Kuehne, Alan D Salama, Sarita Workman, David Wrench, Mohammed Yousuf Karim, Recommendations for the management of secondary hypogammaglobulinaemia due to B cell targeted therapies in autoimmune rheumatic diseases, Rheumatology, Volume 58, Issue 5, May 2019, Pages 889–896
- Emmi, G., Bettiol, A., Gelain, E. et al. Evidence-Based Guideline for the diagnosis and management of eosinophilic granulomatosis with polyangiitis. Nat Rev Rheumatol 19, 378–393 (2023). https://doi.org/10.1038/s41584-023-00958-w
- Greco A, Marinelli C, Fusconi M, et al. Clinic manifestations in granulomatosis with polyangiitis. International Journal of Immunopathology and Pharmacology. 2016;29(2):151-159. doi:10.1177/0394632015617063.
- Hashmi MF, Jain V, Tiwari V. Microscopic Polyangiitis. [Updated 2023 Feb 19]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK531484/
- Greco, Antonio; et al. Microscopic polyangiitis: Advances in diagnostic and therapeutic approaches. Autoimmunity Reviews, 2015.
- Bossuyt, X., Cohen Tervaert, JW., Arimura, Y. et al. Revised 2017 international consensus on testing of ANCAs in granulomatosis with polyangiitis and microscopic polyangiitis. Nat Rev Rheumatol 13, 683–692 (2017). https://doi.org/10.1038/nrrheum.2017.140.
- Moiseev S, Cohen Tervaert JW, Arimura Y , et al. 2020 international consensus on ANCA testing beyond systemic vasculitis. Autoimmun Rev 2020;19:102618. doi:10.1016/j.autrev.2020.102618
- Mariana Freitas de Aguiar, João Gabriel Dantas, Ana Beatriz Bacchiega, Zoraida Sachetto. Métricas nas vasculites sistêmicas. Revista da Sociedade Paulista de Reumatologia, 2022. DOI: https://doi.org/10.46833/reumatologiasp.2022.21.2.59-71